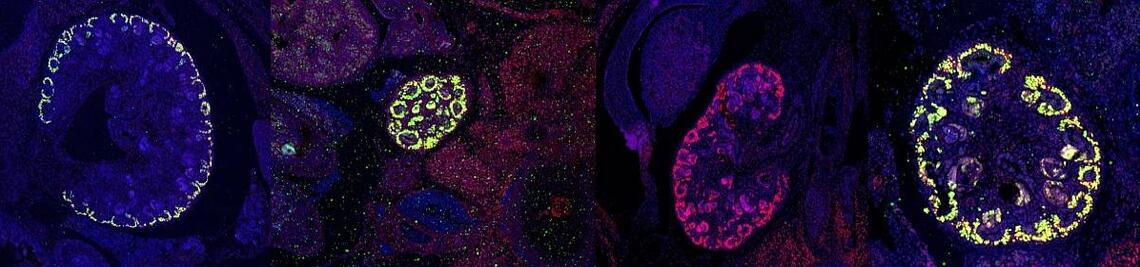
Nierentumor: Genetischer Auslöser entdeckt
18.06.2018Wissenschaftler der Universität Würzburg haben neue molekulare Biomarker für seltene Nierentumoren bei Kleinkindern identifiziert. Diese könnten sich als Angriffspunkte für neue Therapien anbieten.
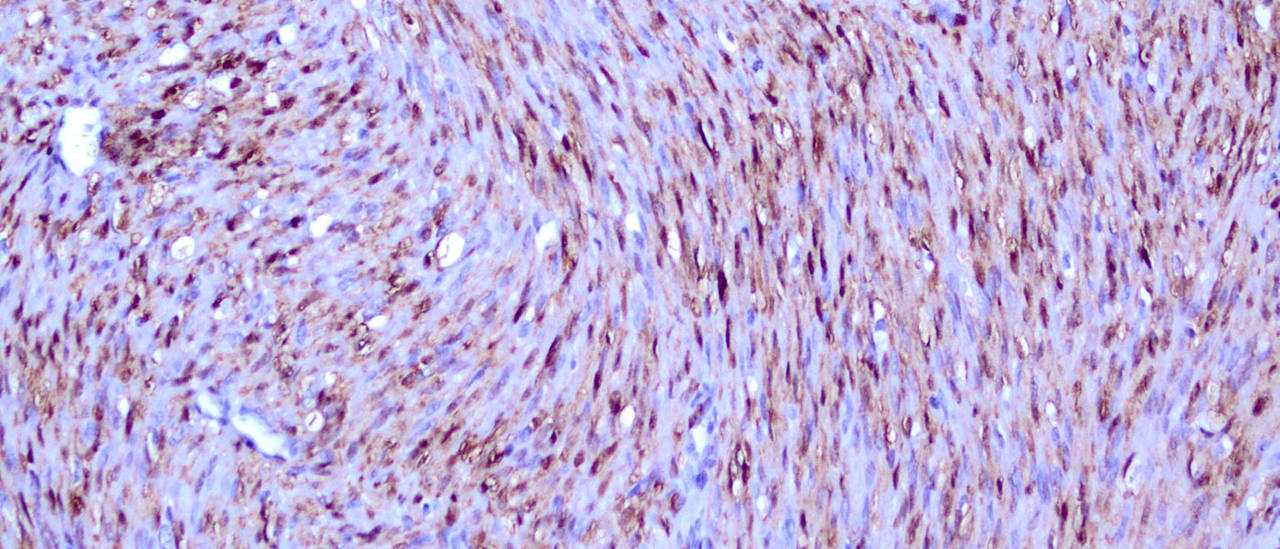
Klassisches mesoblastisches Nephrom mit typischen Bündeln von Spindelzellen und starker Anfärbung für pERK im Zellkern (Bild: Andreas Rosenwald)
Es ist bösartig und kann bereits in den ersten Lebensmonaten von Säuglingen oder sogar schon vor der Geburt auftreten: das congenitale mesoblastische Nephrom (CMN). Glücklicherweise ist der Nierentumor sehr selten und lässt sich oftmals mit einem chirurgischen Eingriff heilen. Weitere spezifische Behandlungsmöglichkeiten existieren jedoch nicht – auch wegen der bislang ungeklärten Ursachen dieses Tumors.
Ein Enzym wird hyperaktiv
Drei Unterarten dieses Nierentumors sind der Wissenschaft bekannt. Für eine dieser Unterarten hat ein internationales Forscherteam jetzt erstmals einen genetischen Auslöser identifiziert. Verantwortlich für diesen Durchbruch waren Jenny Wegert und Professor Manfred Gessler vom Lehrstuhl für Entwicklungsbiochemie am Biozentrum der Julius-Maximilians-Universität Würzburg (JMU); daran beteiligt waren Sam Behjati und Grace Collord vom Wellcome Sanger Centre (Cambridge) und Christian Vokuhl von der Universität Kiel. Die Ergebnisse ihrer Arbeit stellen sie in der aktuellen Ausgabe der Fachzeitschrift Nature Communications vor.
„Anhand von Gewebeschnitten lassen sich beim congenitalen mesoblastischen Nephrom drei Untergruppen definieren: das klassische, das zelluläre und das gemischte Nephrom“, erklärt Manfred Gessler. Für das zelluläre CMN sei seit Langem eine charakteristische Veränderung der Chromosomen als Auslöser bekannt. In diesem Fall fusionieren zwei Gene miteinander, was zu einer überschießenden Aktivität eines Enzyms führt. Für die klassische Variante des CMN waren bislang keine typischen genetischen Veränderungen bekannt.
Neuartige Mutation nachgewiesen
Das hat sich nun geändert: „Durch Genomanalysen von Tumor- und Blutproben der Patienten konnten wir in gut 70 Prozent der klassischen CMN eine neuartige Mutation des Rezeptors für den epidermalen Wachstumsfaktor (EGFR) nachweisen“, schildert Wegert das zentrale Ergebnis der Studie. Die Wissenschaftler entdeckten im Erbgut der betroffenen Zellen eine Verdoppelung der enzymatisch aktiven Kinase-Region. Dies hat zur Folge, dass der EGF-Rezeptor überaktiv wird und die Tumorzellen dauerhaft zum Wachstum angeregt. Wie weitere Untersuchungen zeigten, weist auch die gemischte Variante des Tumors in der Mehrzahl der Fälle eine solche Mutation auf.
Sowohl bei der klassischen als auch bei der zellulären Variante des CMN starten die genetischen Veränderungen einen der wichtigsten Signalwege für die Aktivierung des Zellwachstums: die sogenannte MAP-Kinase-Kaskade. Dabei schalten sie unter anderem die in der Zelle vorliegende BRAF-Kinase an. „Interessanterweise war das Gen für genau dieses Protein auch in einigen der Tumoren mutiert, in denen weder die für die klassische, noch die für die zelluläre Variante verantwortliche Mutation nachgewiesen werden konnte“, erklärt Gessler.
In diesen Fällen kam es im betroffenen Gen zum Verlust einer Region, die für die Hemmung der BRAF-Kinaseaktivität zuständig ist, mit der Folge, dass das Protein daueraktiv ist und die MAP-Kinase-Kaskade angeschaltet bleibt.
Parallelen zu Tumoren bei Erwachsenen
Dem Forscherteam ist es damit gelungen, diesen seltenen Tumor des Säuglingsalters in nahezu allen Fällen auf die Aktivierung eines zentralen Signalweges zurückzuführen, der auch in vielen Tumoren des Erwachsenenalters eine wesentliche Rolle spielt, was Professor Andreas Rosenwald vom Institut für Pathologie der JMU durch entsprechende Färbungen an Tumorschnitten untermauern konnte.
Insbesondere für CMN-Patienten, die chirurgisch nicht ausreichend behandelt werden können, liefern diese Erkenntnisse jetzt möglicherweise neue Behandlungsansätze durch die Übertragung bewährter Therapieprinzipien der Erwachsenenonkologie auf die Therapie des congenitalen mesoblastischen Nephroms.
Recurrent intragenic rearrangements of EGFR and BRAF in soft tissue tumors of infants. Nature Communications, doi: 10.1038/s41467-018-04650-6, https://www.nature.com/articles/s41467-018-04650-6 .
Kontakt
Prof. Dr. Manfred Gessler, Lehrstuhl Entwicklungsbiochemie
T: +49 931 31 84159 / 84160 (Sekr.), gessler@biozentrum.uni-wuerzburg.de
Von Gunnar Bartsch