Prof. em. Müller-Reible

Prof. em. Dr. rer. nat. Clemens R. Müller-Reible
E-Mail: crm@biozentrum.uni-wuerzburg.de
Tel.: +49-931-31-84063
Fax: +49-931-31-84069
Vita
Clemens R. Müller-Reible obtained his degree in biology (Diplom-Biologe) from the university of Freiburg. Following his Ph.D. in biochemistry he has worked from 1978 to 1983 as a post-doc with Hans-Hilger Ropers at the department of human genetics and anthropology at Freiburg university. In 1983 he was appointed as lecturer at the newly founded department of human genetics of Würzburg university. There he founded a laboratory for human molecular genetics and clinical genetic diagnostics. He obtained his habilitation in human genetics from the medical faculty of Würzburg university in 1987 and was appointed Associate Professor in 1996.
Since 1998 he serves as a recognized expert inspector for human molecular genetics for the Deutsche Akkreditierungsstelle. In 2009 he was appointed by the Federal Ministry of Health and Social Affairs to the German Gene Diagnostics Commission at the Robert-Koch-Institut, Berlin.
Malignant Hyperthermia
Malignant Hyperthermia (MH) is the term used to describe a crisis of calcium metabolism which can be triggered by halogenated anesthetic gases like halothane or by muscle relaxants like succinyl choline. Genetically predisposed individuals do not present any symptoms in daily life. Following an exposue, however, they may develop acute and life-threatening symptoms including generalized rigor, high fever, tachycardia, hypocapnia and eventually multi-organ failure.
The genetic basis of MH are mutations in the major calcium channel of skeletal muscle, the ryanodine receptor (RYR1). Upon stimulation, the membrane depolarisation is sensed by the voltage-sensitive DHP receptor which causes RYR1 by direct physical interaction to open and release Ca2+ from intracellular stores in the sarcoplasmic reticulum.
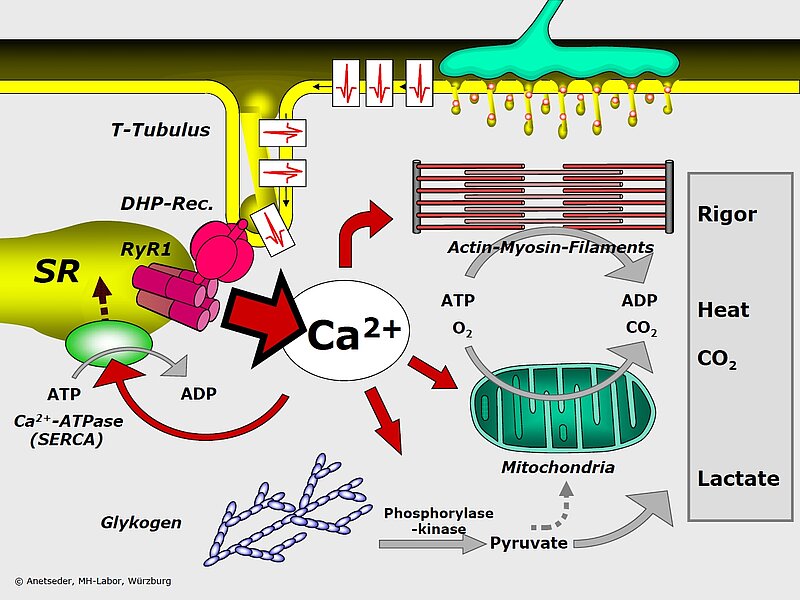
In collaboration with the MH-laboratory of the department of anesthesiology of Würzburg university hospital and with the European Malignant Hyperthermia Group we and others have shown that MH is doubly heterogeneous: there may be as many as 5 MH loci in our genome and more than 200 mutations have as yet been identified in the major locus MHS1 (= RYR1). Amino acid substitutions are the prevalent mutation type conferring a dominant functional alteration on RYR1.
At the same time, RYR1 is the major gene for congenital myopathies without CNS involvement, such as Central Core Disease, Multi-Minicore Disease, centronuclear myopathy, fibre-type-disproportion. In contrast to MH, these disorders are mostly transmitted in an autosomal recessive mode. Compound heterozygosity for a truncation mutation (stop or frameshift) and a missense-mutation is frequently found. Furthermore, since RYR1 acts as a homotetrameric protein, ist function is dose-dependent. Therefore, a reduced expression level of RYR1 in skeletal muscle can lead to functional impairment.
Selected publications
Broman M, Kleinschnitz I, Bach JE, Rost S, Islander G, Müller CR. Next-generation DNA sequencing of a Swedish malignant hyperthermia cohort. Clin Genet. 2015 Oct;88(4):381-5.
Glahn KP, Ellis FR, Halsall PJ, Müller CR, Snoeck MM, Urwyler A, Wappler F; European Malignant Hyperthermia Group. Recognizing and managing a malignant hyperthermia crisis: guidelines from the European Malignant Hyperthermia Group. Br J Anaesth. 2010.
Zhou H, Jungbluth H, Sewry CA, Feng L, Bertini E, Bushby K, Straub V, Roper H, Rose MR, Brockington M, Kinali M, Manzur A, Robb S, Appleton R, Messina S, D'Amico A, Quinlivan R, Swash M, Müller CR, Brown S, Treves S, Muntoni F. Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 2007.
Ducreux S, Zorzato F, Ferreiro A, Jungbluth H, Muntoni F, Monnier N, Muller CR, Treves S. Functional properties of ryanodine receptors carrying 3 amino acid substitutions identified in patients affected by multi-minicore disease and central core disease, expressed in immortalised lymphocytes. Biochem J. 2006.
Anetseder M, Hager M, Müller CR, Roewer N. Diagnosis of susceptibility to malignant hyperthermia by use of a metabolic test. Lancet 2002.
Jungbluth H, Müller CR, Halliger-Keller B, Brockington M, Brown SC, Feng L, Chattopadhyay A, Mercuri E, Manzur AY, Ferreiro A, Laing NG, Davis MR, Roper HP, Dubowitz V, Bydder G, Sewry CA, Muntoni F. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology 2002.
Quality assurance of clinical genetic diagnostics
As a clinical diagnostic field, nucleic acid analysis is a relatively young discipline. Since the introduction of the basic methods (Sanger et al. 1977; Mullis et al. 1985) the technologies are undergoing a continuous development. The level of technical standarisation is low and the availability of FDA- or CE-approved diagnostic kits for genetics is limited.
On the other hand, a genetic diagnosis is usually made only once in the lifetime of a patient and has immediate implications for his/her family members. This calls for the highest possible accuracy and reliability of genetic testing methods. The tools for measuring and improving such standards are internal and external quality assurance (QA).
In the early 1990ies, the community of geneticists in Germany has started to set up a program of external QA by the exchange of anonymised DNA samples. Coordinated from Würzburg, this has evolved in the course of 15 years into an EQA program covering 15 heritable disorders and serving over 250 participants per year. However, coordinating and running these EQA schemes on a voluntary basis became increasingly difficult. Therefore, in 1998 the EU-funded project "European Molecular Genetics Quality Network (EMQN)" came just in time. In fact, EMQN turned out to be one of the very few projects to live on after EU-support had ended. Based on subscription by the participants, EMQN is still going strong and has expanded its menu considerably (www.emqn.org).
Not too surprisingly perhaps, the German and the European approaches to EQA in molecular genetics shared many conceptual and organisational details. With the growing complexity of testing and the increasing difficulties in recruiting scheme organisers and EQA materials, a convergence of activities was more than suggestive. Therefore, in 2006 GfH and EMQN have signed an agreement of cooperation in order to jointly run the EQA programs.
In parallel to the development of EQA programs, the publication of best practice guidelines for molecular genetic testing became an important area of activity.
Selected publications
Abbs S, Tuffery-Giraud S, Bakker E, Ferlini A, Sejersen T, Mueller CR. Best Practice Guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscular Disorders 2010.
Mattocks CJ, Morris MA, Matthijs G, Swinnen E, Corveleyn A, Dequeker E, Müller CR, Pratt V, Wallace A. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur J Hum Genet 2010.
McGovern MM, Elles R, Beretta I, Somerville MJ, Hoefler G, Keinanen M, Barton D, Carson N, Dequeker E, Brdicka R, Blazkova A, Aymé S, Schnieders B, Muller CR, Dalen V, Martinez AA, Kristoffersson U, Ozguc M, Mueller H, Boone J, Lubin IM, Sequeiros J, Taruscio D, Williamson B, Mainland L, Yoshikura H, Ronchi E. Report of an international survey of molecular genetic testing laboratories. Community Genet 2007.
Müller CR, Kristoffersson U, Stoppa-Lyonnet D. External quality assessment for mutation detection in the BRCA1 and BRCA2 genes: EMQN's experience of 3 years. Ann Oncol 2004.
Simoni M, Bakker E, Eurlings MC, Matthijs G, Moro E, Müller CR, Vogt PH. Laboratory guidelines for molecular diagnosis of Y-chromosomal microdeletions. Int J Androl 1999.