Humangenetik
Zum 1. Oktober 2025 wurde der Fachbereich Humangenetik der Julius-Maximilians-Universität Würzburg durch das Ministerium für Wissenschaft und Kunst auf das Universitätsklinikum Würzburg (UKW) übertragen und wird dort als Institut für Klinische Genetik und Genommedizin (KGGM) fortgeführt. Frau Prof. Dr. med. Anke Katharina Bergmann hat die Leitung des Institutes am UKW übernommen.
Mit dieser Veränderung ist auch eine Erweiterung unseres Leistungsspektrums verbunden, um Ihnen und Ihren Patientinnen und Patienten künftig ein noch umfassenderes Angebot bieten zu können. Unser diagnostisches und therapeutisches Spektrum stellen wir auf unserer neuen Homepage vor (https://www.ukw.de/klinische-genetik-und-genommedizin/startseite/).
Für eine humangenetische Beratung kann der Überweisungsschein Muster 6 verwendet werden. Termine für Patienten können auch online über unsere Homepage vereinbart werden.
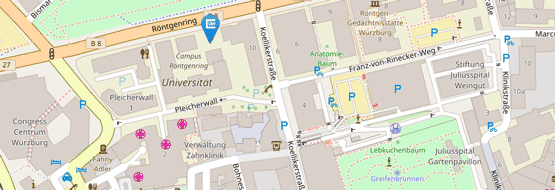
Für Patientenvorstellungen befindet sich die Hochschulambulanz des Institutes auf dem UKW-Gelände:
Haus C13
Josef-Schneider-Str. 2
97080 Würzburg
(https://www.ukw.de/klinische-genetik-und-genommedizin/humangenetische-beratung/)
Ergänzend dazu möchten wir Sie darüber informieren, dass ab dem 1. Januar 2026 auch Einsendungen in unsere Hochschulambulanz für Humangenetik möglich sind. Sie können damit Probeneinsendungen mit Laborüberweisungsschein Muster 10 direkt an uns senden.
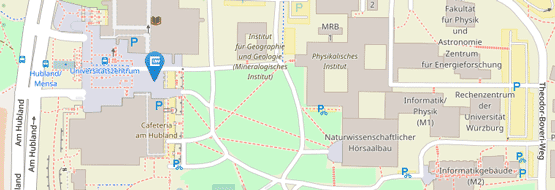
Die Adresse für die Probenannahme bleibt weiterhin unverändert:
Institut für Klinische Genetik und Genommedizin
Biozentrum
Am Hubland
97074 Würzburg
Einsendeformular, GenDG-Einwilligung etc. finden Sie ab dem 1. Januar 2026 auf unserer Homepage im Bereich „Diagnostik“
(https://www.ukw.de/klinische-genetik-und-genommedizin/diagnostik/)
Wir sind weiterhin zuverlässig für Sie erreichbar und setzen uns dafür ein, die Qualität unserer Arbeit kontinuierlich zu steigern und die Bearbeitungszeiten zu optimieren.