ANTIGENIC VARIATION
Antigenic variation (Engstler lab)
Trypanosomes change their wardrobe.
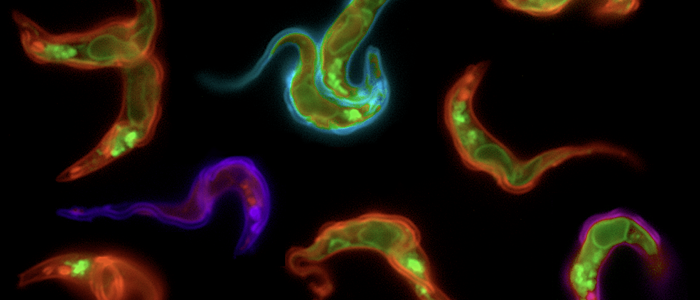
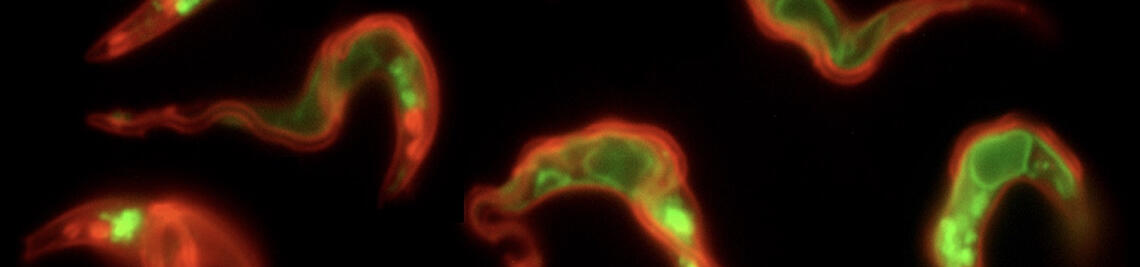
The parasites posses hundreds of coats.
The VSG expression site is multifunctional.
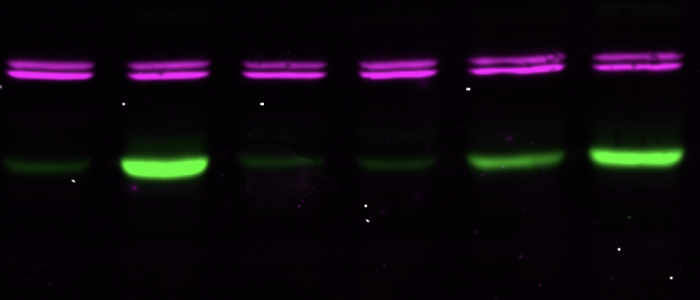
It is the trypanosome's virulence hub.
SIF is not the only way to become stumpy.
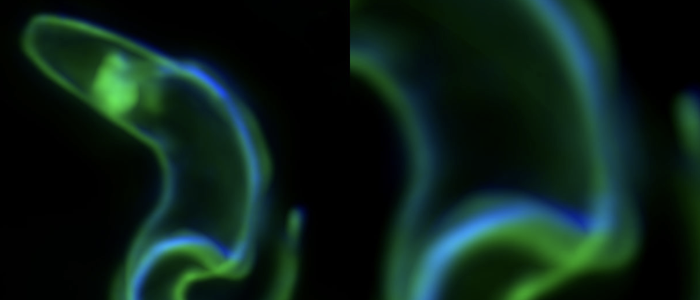
It's generally thought that SIF-induced stumpy formation is essential for the parasites' life cycle.
Antigenic variation ...
Pathogenic bacteria and protozoan parasites often employ a coat of surface molecules to protect themselves from host immune attack. These surface coats are generally variable and hence, not only act as physical shield but have evolved as efficient camouflage strategy. The surface-exposed proteins are mostly members of large families and are subject to antigenic variation, i.e. they are sporadically exchanged. This allows the persistence of the pathogens in the host, as well as reinfection. An extensively studied model for antigenic variation is the protozoan parasite Trypanosoma brucei.The trypanosome surface coat consists of millions of identical copies of a variant surface glycoprotein (VSG). The highly immunogenic VSGs cause a rapid host immune response, which is thought to lead to an almost complete elimination of the parasite population. Only parasites that have switched to the expression of an immunologically distinct VSG survive. Thus, at any given time just one VSG out of a repertoire of several hundreds of VSG genes is expressed and dominates the cell surface of the pathogen. Trypanosoma brucei exploits both, genetic and epigenetic mechanisms for antigenic variation. Allelic exclusion, which may be achieved by epigenetic modifications, ensures that only one VSG gene is expressed from one of 15 telomeric expression sites (ES). The open chromatin structure of the active ES is thought to facilitate its transcription by RNA polymerase I in a distinct extranucleolar compartment termed the expression site body (ESB). The large repertoire of silent VSG copies is subject to frequent rearrangements, resulting in the continuous production of new mosaic variants. A VSG switch is recombinatorial when the actively transcribed VSG gene is replaced by another variant. Besides by gene conversion, antigenic variation can occur by telomere exchange, i.e. by recombinatorial cross-over of chromosome ends. Alternatively, the expressed VSG can be exchanged by transcriptional silencing of the active ES and activation of another, previously non-transcribed ES promoter. This so-called 'in situ' switch does not involve genetic recombination but possibly epigenetic modifications. We have worked on an interesting new facet of 'in situ' switching.
... and development?
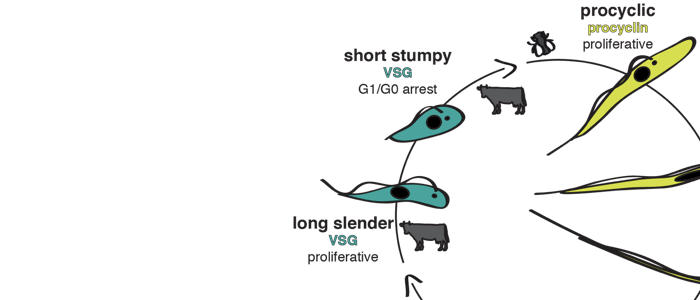
About 20 years ago a quorum sensing pathway was discovered that controls development of the African sleeping sickness pathogen Trypanosoma brucei. The parasites secrete the (still elusive) ‘stumpy induction factor’ (SIF) that drives proliferating slender trypanosomes into the cell cycle arrested stumpy stage. In this way, the parasites limit their cell density and hence, the burden they impose on their mammalian host. It has further been proposed that only SIF-induced stumpy trypanosomes can successfully infect the transmitting insect vector, the tsetse fly. We have shown that this is not the case and have proposed an alternative route to the stumpy stage and the tsetse fly. In fact, the ‘stumpy induction factor’ is not required for developmental progression. Furthermore, we suggest that ‘errors’ in trypanosome antigenic variation are not necessarily lethal for the parasites, as they can lead to stumpy development. We propose that the impact of ‘unsuccessful’ antigenic switches on parasite differentiation is very significant and so far unexplored. Moreover, our studies emphasize the importance of studying natural (primary) parasite strains. We have used pleomorphic parasites and discovered a striking phenotypic plasticity: The trypanosomes can respond to an erroneous antigenic VSG switch by becoming stumpy (and hence tsetse-infective) or they can halt the switch event and thus, gain time for correcting the error. Our work has shed new light on trypanosome antigenic variation and development, as it establishes the VSG expression site as a stochastic switch controlling virulence and persistence. This further promotes African trypanosomes as versatile model parasites.
Select publications
Bartossek T, Jones NG, Schäfer C, Cvitković M, Glogger M, Mott HR, Kuper J, Brennich M, Carrington M, Smith A-S, Fenz S, Kisker C, Engstler M. (2017) Structural basis for the shielding function of the dynamic trypanosome VSG coat. Nature Microbiology, 2017 Sep 11. doi: 10.1038/s41564-017-0013-6.[Epub ahead of print] PubMed PMID: 28894098.
Zimmermann, H., Subota, I.; Batram, C, Kramer, S, Janzen, C, Jones, N, Engstler, M (2017) A Quorum Sensing-independent Path to Stumpy Development in Trypanosoma brucei. Plos Pathogens doi: 10.1371/journal.ppat.1006324
Dejung M, Subota I, Bucerius F, Dindar G, Freiwald A, Engstler M, Boshart M, Butter F, Janzen CJ. (2016) Quantitative Proteomics Uncovers Novel Factors Involved in Developmental Differentiation of Trypanosoma brucei. PLoS Pathog. doi: 10.1371/journal.ppat.1005439. PMID: 26910529
Batram, C., Jones, N.G., Janzen, C.J., Markert, S.M., and Engstler, M. (2014). Expression site attenuation mechanistically links antigenic variation and development in Trypanosoma brucei. Elife 3, e02324.